

Abstract
Background:
Sickle cell disorder (SCD) and transfusion dependent beta-thalassemia (TDT) are inherited disorders of β-globin genes leading to ineffective erythropoiesis, hemolysis and multi-organ damage. Though allogenic hematopoietic cell transplantation (allo-HCT) in the only potentially curative therapy, lack of full HLA-matched donor, increased risk of myeloablative conditioning (MAC) regimen related toxicity, poor prognosis in older age groups (>14 years), and a high risk of infections and graft versus host disease (GVHD) culminating into a high transplant related mortality (TRM) preclude HCT as a treatment option for many patients.
Gene therapy has been proposed as a relatively new treatment option for these monogenic disorders. It involves transfection of harvested CD34+ hematopoietic stem cells (HSC) with viral vector to transfer a normal beta globin gene, which are then transplanted back to the autologous donor. However, the available data on gene therapy is extremely variable. The purpose of this review is to evaluate the risks and outcomes of SCD and TDT patients undergoing gene therapy.
Methods:
A systematic search of databases using PubMed, Embase, Web of science, Cochrane database and Clinicaltrials.gov was performed on with no restrictions of language or time period. A total of 4312 articles were identified and after duplicates removal, 3508 articles were screened for relevance. Clinical studies reporting the most recent follow up results about the efficacy and safety of gene therapy in patients with hemoglobinopathies were considered for inclusion. Their quality assessment was done using the Cochrane tool, and bias was evaluated regarding the study selection, performance, detection, attrition and the reporting of the clinical trials.
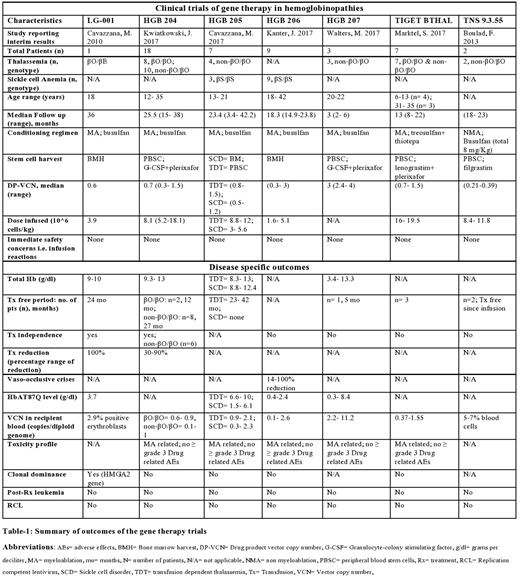
RESULTS (table-1):
Only 7 articles qualified for the strict selection criteria with an N of 47: six studies were phase I trials (Cavazzana, M. et al 2010, Boulad, F. et al 2013, Marktel, S. et al 2017, Kwiatkowski, J. et al 2017, Cavazzana, M. et al 2017, Kanter, J. et al 2017) and one was a phase III trial (Walters, M. et al 2017). Thirty five patients (74.4%) had TDT while 12 patients (25.5%) had SCD. All these patients did not have a suitable HLA-matched HCT donor. The length of follow up was heterogeneous owing to the step wise enrollment of participants and ranged from 2-36 months. Except for 4 cases, the age of included population was >13 years. Lentivirus BB305 encoding the HbAT87Q globin gene was used for gene delivery in 81% patients (n=38). The normal beta globin gene was transduced in remaining 9 TDT cases and it involved transduction using TNS 9.3.55 vector in two cases and with GLOBE lentivirus in 7 cases. MAC with busulfan was used in 81% cases (n=38), while treosulfan+thiotepa was used in 7 cases. The non-myeloablative (NMA) conditioning with busulfan was used in only 2 cases (both TDT). Bone marrow harvest was the source of HSCs in all SCD cases. Except for 3 SCD cases in HGB-204 trial and 3 TDT cases in HGB-207 which measured the potential improvement in outcome by an increase in DP-VCN, the range of drug vector copy number (DP-VCN) was 0.3-1.5 copies per diploid genome. The total hemoglobin (HB) level at last follow up was reported for 62% cases (n=29) and it was >8.8 g/dl in all cases. Twenty-one (60%) thalassemia cases were in a transfusion (Tx) free state at last follow up, including the 7 cases (20%) who developed a complete independence from transfusions. No such Tx-free state was noted in SCD cases although they developed a reduction in the Tx-requirement and 75% SCD cases (n=9) had a 30-100% reduction in the frequency of VOCs. All the cases were alive at their recent follow up with manageable toxicity form conditioning. Acceptable safety until the median follow-up from the 7 trials was established in all cases, with no reported development of post-treatment leukemia or new malignancy in these trials.
DISCUSSION:
In this largest series of gene therapy for the treatment of hemoglobinopathies (to our knowledge), we find it to be clinically effective in terms of reduction in transfusion needs; data to suggest an improved OS and disease free survival is promising, however long term data is awaited. Gene therapy appears to bypass the limitations faced by allo-HCT especially a high early TRM. Randomized trials reporting the efficacy and safety outcomes with longer follow ups are urgently required.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal